Reaktion als Säure Q1
Die Carboxygruppe oder kurz COOH-Gruppe ist in der Lage, ein Proton abzugeben. Alle Carbonsäuren sind daher Brönsted-Säuren.
Betrachten wir einmal die Säurestärken verschiedener Carbonsäuren[3].
| Säure | pKS-Wert |
| Methansäure (Ameisensäure) | 3,77 |
| Ethansäure (Essigsäure) | 4,76 |
| Propansäure | 4,87 |
| Butansäure (Buttersäure) | 4,82 |
| Pentansäure (Valeriansäure) | 4,84 |
Wie es aussieht, ist Methansäure die stärkste Carbonsäure und Ethansäure die zweitstärkste. Die folgenden Glieder der homologen Reihe der Monocarbonsäuren unterscheiden sich nicht mehr wesentlich in ihrer Säurestärke. Außerdem nimmt die Wasserlöslichkeit der Carbonsäuren mit jedem zusätzlichen C-Atom ab, so dass der Säurecharakter dann keine Rolle mehr spielt.
Vergleichen wir einmal die Ethansäure mit dem Ethanol. Auch Ethanol hat eine OH-Gruppe, könnte also theoretisch ein H-Atom als Proton abgeben und damit als Säure wirken.
Theoretisch ist das auch möglich, allerdings beträgt der pKS-Wert von Ethanol sage und schreibe 16, unter normalen Bedingungen fällt einem Ethanol-Molekül also nicht im Traum ein, ein Proton an eine Base abzugeben. Aber vor Überraschungen ist man ja in der organischen Chemie nicht gefeit. Wer hätte zum Beispiel gedacht, dass Aceton-Moleküle (pKS-Wert ca. 20, also noch 10.000 mal schwächer als Ethanol) durchaus ein Proton an eine Base abgeben können. Genau das passiert nämlich bei der Synthese von Dibenzalpropanon, wie ich auf der entsprechenden Seite zur nucleophilen Addition ausgeführt habe.
Wieso hat Ethansäure einen so viel geringeren pKS-Wert als Ethanol, ist also eine viel stärkere Säure?
Der Hauptgrund wird sicherlich das zusätzliche Sauerstoff-Atom der Carboxygruppe sein. Um zu verstehen, warum die Carboxygruppe ihr Proton viel leichter abgibt als eine einfache Hydroxygruppe, betrachten wir einmal eine Zeichnung des Carboxylat-Ions:
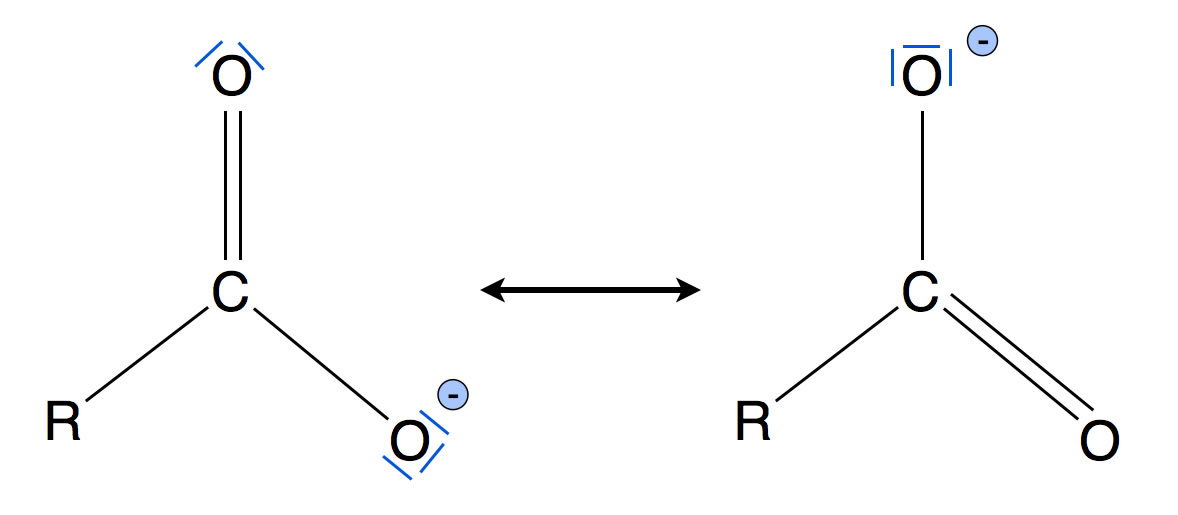
Das Carboxylat-Ion hat zwei Grenzstrukturen
Das Carboxylat-Ion kann in zwei verschiedenen Grenzstrukturen vorkommen. Die tatsächliche ("wahre") Struktur des Carboxylat-Ions liegt irgendwo dazwischen. Man kann auch sagen, dass das Ion in beiden Strukturen gleichzeitig vorkommt. Veraltet ist dagegen die Hypothese, nach der das Ion ein paar Tausend Mal pro Sekunde die Strukturen wechselt.
Die negative Ladung befindet sich weder an dem einen O-Atom noch an dem anderen O-Atom, sondern an beiden O-Atomen gleichzeitig. Man sagt, die negative Ladung ist delokalisiert.
Nun gibt es eine gute alte Regel in der organischen Chemie, die man sich gut merken sollte:
Je mehr Grenzstrukturen es von einer Verbindung gibt, desto stabiler ist diese.
Das Carboxylat-Ion kommt in zwei Grenzstrukturen vor, von der Carboxygruppe gibt es aber nur eine mögliche Struktur. Das Carboxylat-Ion ist also demnach stabiler als die Carbonsäure. Aus diesem Grund kann das H-Atom der Carboxygruppe leicht abgegeben werden, die Bildung des Carboxylat-Ions ist energetisch sehr günstig[1].
Gibt ein Alkohol ein Proton ab, so gibt es nur eine Möglichkeit, das Alkoholat-Ion zu bilden. Das Alkoholat-Ion ist also nicht energetisch günstiger als das Alkohol-Molekül. Aus diesem Grund wird ein Alkohol also nur dann ein Proton abgeben, wenn eine wirklich extrem starke Base anwesend ist, während bei Essigsäure und anderen Carbonsäure bereits die Anwesentheit von Wasser-Molekülen ausreicht, um Protonen abzugeben.
Salze der Carbonsäuren
Gibt man eine wasserlösliche Carbonsäure mit einer Lauge zusammen, so bilden sich Carbonsäuresalze. Aus Essigsäure und Natronlauge beispielsweise bildet sich Natriumacetat, aus Kalilauge und Ameisensäure Kaliumformiat. Die Namen der Säurereste stehen oft in keiner Beziehung zu den Trivialnamen oder auch zu den IUPAC-Namen der entsprechenden Säure. Daher hier eine kleine Tabelle mit den wichtigsten Säurerest-Namen.
| Säure | Name des Restes |
| Methansäure (Ameisensäure) | Formiat |
| Ethansäure (Essigsäure) | Acetat |
| Propansäure | Propionat |
| Butansäure (Buttersäure) | Butyrat |
| Pentansäure (Valeriansäure) | Valerat |
Substitution der Hydroxygruppe EF, Q1
Kommen wir nun zu den "richtigen" chemischen Reaktionen der Carbonsäuren. Eine der wichtigsten Reaktionstypen ist die Substitution (Ersetzung) der OH-Gruppe. Hierbei handelt es sich um eine nucleophile Substitution, denn das C-Atom der Carboxygruppe ist ja stark positiv polarisiert.
Bildung von Säurechloriden Q1
Die Bildung von Säurechloriden ist sicherlich die wichtigste Substitutionsreaktion der Carbonsäuren. Säurechloride sind sehr reaktive Derivate der Carbonsäuren und dienen zur Synthese vieler anderer wichtiger Verbindungen, beispielsweise von Estern oder Amiden.
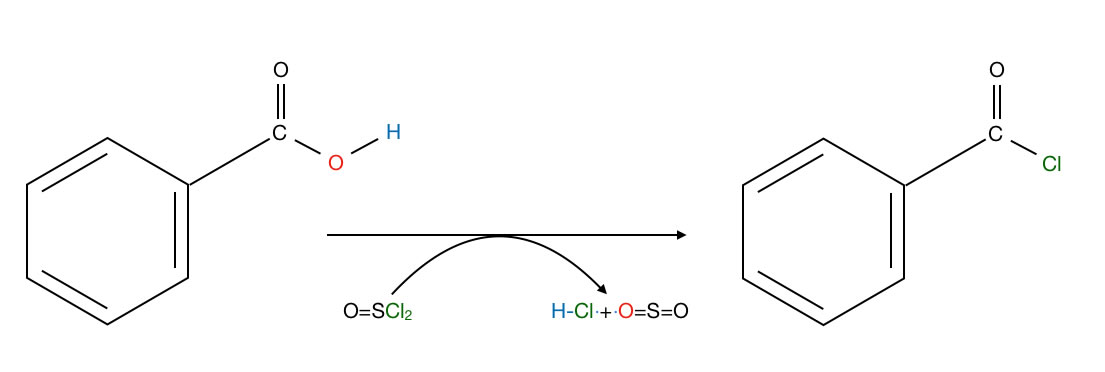
Bildung von Benzoylchlorid aus Benzoesäure und Thionylchlorid
Kocht man Benzoesäure mit Thionylchlorid (Kolben mit Rückflusskühler), so erhält man das Säurechlorid Benzoylchlorid. Die Verbindung Thionylchlorid ist für diese Synthese besonders gut geeignet, weil ausschließlich gasförmige Nebenprodukte (Chlorwasserstoff und Schwefeldioxid) entstehen, die leicht vom eigentlichen Reaktionsprodukt abgetrennt werden können.
Alternativ kann man Säurechloride auch mit Hilfe von Phosphortrichlorid PCl3 oder Phosphorpentachlorid PCl5 herstellen[1].
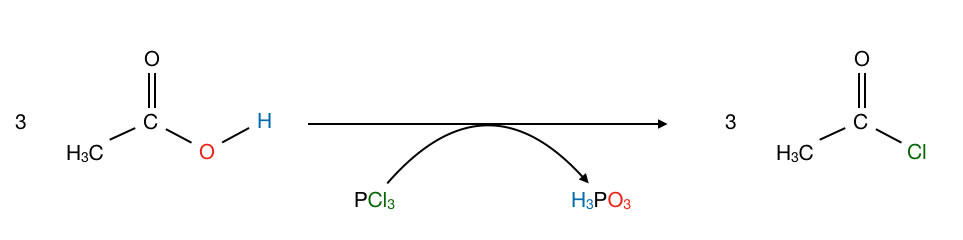
Bildung von Essigsäurechlorid aus Essigsäure mit Hilfe von Phosphortrichlorid
Bildung von Estern EF, Q1
Auch die Bildung eines Esters aus einer Carbonsäure und einem Alkohol ist eine nucleophile Substitution. Den Reaktionsmechanismus der säurekatalysierten Estersynthese finden Sie auf einer eigenen Seite. Leider ist die Umsetzung einer Carbonsäure mit einem Alkohol eine Gleichgewichtsreaktion, die Ausbeute ist nicht immer so hoch wie man es sich wünscht, weil sich das chemische Gleichgewicht schon dann einstellt, wenn erst Teil der Carbonsäure reagiert hat. Bei der Umsetzung von Essigsäure mit Ethanol kommt die Reaktion zum Beispiel schon dann zum (scheinbaren) Stillstand, wenn ca. 2/3 der Säure bzw. des Alkohols reagiert haben.
Lässt man dagegen ein Säurechlorid mit dem Alkohol reagieren, liegt das Gleichgewicht der Reaktion wegen der hohen Reaktivität des Säurechlorides fast ausschließlich auf der Produktseite, also auf der Seite des gewünschten Esters. Genau genommen sind beide Schritte der Estersynthese nahezu irreversibel, sowohl die Bildung des Säurechlorids wie auch die Bildung des Esters aus dem Säurechlorid.
Der Nachteil der Estersynthese über ein Säurechlorid ist, dass es sich um zwei Reaktionsschritte handelt. Darum versucht man viele Ester direkt herzustellen, also aus der Carbonsäure und dem Alkohol. Eine höhere Ausbeute an Ester erzielt man durch gezielte Verschiebung des chemischen Gleichgewichtes bei der Reaktion. Damit wären wir bei einem Thema, das für Schüler der Stufe EF interessant ist, denn hier wird das Prinzip des kleinsten Zwanges im Kernlehrplan thematisiert.
Gleichgewichtsverschiebungen bei der Veresterung EF
Für Schüler aus der Stufe EF ist sicherlich interessant, wie man das chemische Gleichgewicht bei der direkten Veresterung (also der Reaktion der Carbonsäure mit dem Alkohol unter Wasserabspaltung) zugunsten des Esters verschieben kann.
Morrison/Boyd führt ein interessantes Beispiel an[1]: Wir wollen einen Ester herstellen. Dazu benötigen wir einen Alkohol und eine Carbonsäure. Angenommen, der Alkohol ist viel billiger als die Carbonsäure. Dann setzen wir einfach die drei-, vier- oder fünffache Menge des Alkohols ein, als eigentlich nötig wäre. Auf diese Weise verschiebt sich das Gleichgewicht der Reaktion weiter auf die rechte Seite, und wir erhalten eine höhere Ausbeute. Ist dagegen die Säure billiger als der Alkohol, machen wir es genau umgekehrt: Säureüberschuss.
"For example, it is worthwhile to use eight moles of cheap ethyl alcohol to convert one mole of valuable gamma-phenylbutyric acid more completely into the ester"[1].
Übersetzung:
"Beispielsweise lohnt es sich, 8 mol des billigen Ethanols anzwenden, um 1 mol der wertvollen gamma-Phenylbuttersäure vollständiger in den Ester umzuwandeln".
Bildung von Amiden Q1
Amide sind Carbonsäure-Derivate, bei denen die OH-Gruppe der Carboxygruppe durch eine NH2-Gruppe (Aminogruppe) ersetzt wurde. Im Allgemeinen werden Amide nicht direkt aus der Carbonsäure und Ammoniak NH3 hergestellt, sondern aus Säurechloriden und Ammoniak oder Aminen (Amine sind Derivate des Ammoniaks, bei denen ein oder mehrere H-Atome durch Alkylgruppen ersetzt wurden).
Reduktion zu Alkoholen Q1
Da man Alkohole meistens leichter synthetisieren kann als eine Carbonsäure, wird oft der umgekehrte Weg gegangen: Aus dem Alkohol wird durch Oxidation eine Carbonsäure hergestellt. Es gibt aber auch Fälle, in denen die Carbonsäure leichter gewonnen werden kann als der entsprechende Alkohol, das ist zum Beispiel bei den langkettigen Fettsäuren der Fall, die man leicht aus Fetten oder Ölen gewinnen kann.
Das beliebteste Reduktionsmittel, mit dem man aus einer Carbonsäure einen primären Alkohol herstellen kann, ist das Lithiumaluminiumhydrid LiAlH4[1,4]. Der genaue Verlauf der Reduktion ist hier jetzt nicht relevant (Mechanismus siehe Chemgapedia), auf jeden Fall entsteht dann unter recht milden Bedingungen der passende primäre Alkohol in hoher Ausbeute aus der Carbonsäure. Allerdings ist das Lithiumaluminiumhydrid recht teuer, daher wird es nur bei der Synthese von Arzneimitteln oder Hormonen eingesetzt[1].
Billiger ist die zweistufige Reduktion: Zunächst wird die Carbonsäure verestert, und dann wird der Ester zu zwei Alkoholen reduziert.
Halogenierung am alpha-C-Atom Q1
Kommen wir nun zu einer ganz anderen Reaktion, bei der die Carboxygruppe zwar eine Rolle spielt, aber nicht an der Reaktion teilnimmt. Das der Carboxygruppe direkt benachbarte C-Atom der Carbonsäure wird als alpha-C-Atom bezeichnet. Die H-Atome, die an diesem C-Atom sitzen, heißen dann entsprechende alpha-H-Atome.
Diese alpha-H-Atome können nun recht leicht durch Halogen-Atome ersetzt werden, vor allem durch Brom oder Chlor. Kleine Mengen an Phosphor dienen dabei als Katalysator. Auf diese Hell-Volhard-Zelinsky-Reaktion soll aber an dieser Stelle nicht weiter eingegangen werden. Es sei nur gesagt, dass sie sehr spezifisch und sehr schnell verläuft und damit eine große Bedeutung für die organische industrielle Synthese hat[4].
Alkylierung am alpha-C-Atom Q1
Starke Basen entziehen einer Carbonsäure nicht nur das H-Atom der Carboxygruppe, sondern darüber hinaus auch noch ein H-Atom des alpha-C-Atoms:
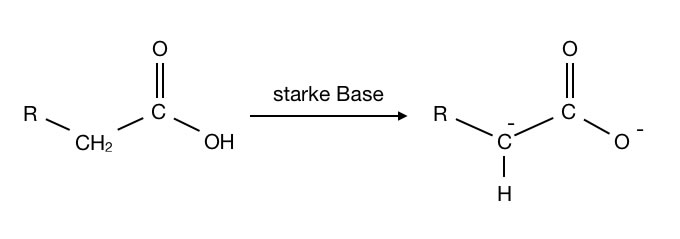
Zwei Moleküle einer starken Base können einer Carbonsäure auch ein alpha-H-Atom entreißen
An das negativ geladene C-Atom kann nun leicht ein elektrophiles Carbenium-Ion angreifen, wie sie zum Beispiel beim ersten Schritt der SN1-Reaktion entstehen oder bei der Addition eines Protons an die C=C-Doppelbindung.
Decarboxylierung Q1
Manche Carbonsäuren geben ihre Carboxygruppe recht leicht in Form von Kohlendioxid ab, man spricht dann von einer Decarboxylierung. Das Ergebnis einer solchen Decarboxylierung ist dann ein einfaches Alkan. Jedenfalls dann, wenn eine Monocarbonsäure decarboxyliert wurde. Ganz einfach ist das bei Monocarbonsäuren wie Essigsäure oder Buttersäure allerdings nicht. Viel besser eigenen sich beta-Ketosäuren und 1,3-Dicarbonsäuren zur Decarboxylierung[4].
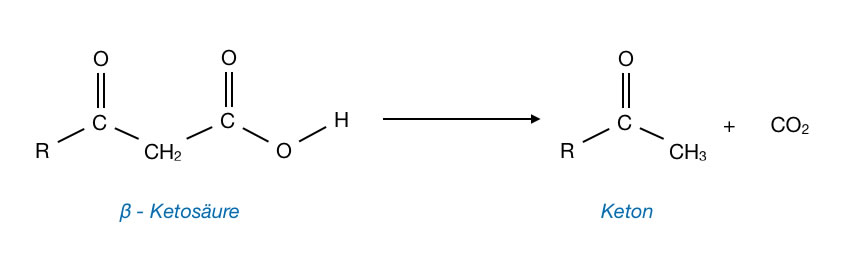
Decarboxylierung einer beta-Ketosäure zu einem Keton
Bei einer beta-Ketosäure ist R eine Alkylgruppe. Ist R dagegen eine Hydroxygruppe, so hat man die 1,3-Dicarbonsäure namens Malonsäure. Malonsäure lässt sich in wässriger Lösung bereits bei 70 ºC decarboxylieren[5].
Substitution der Carboxygruppe Q1
Bei der Decarboxylierung wird die gesamte Carboxygruppe als CO2 abgespalten. Man kann die Carboxygruppe aber auch durch eine andere funktionelle Gruppe ersetzen. Bei der Hunsdiecker-Reaktion wird die COOH-Gruppe zum Beispiel durch ein Halogen-Atom ersetzt. Aus der Carbonsäure erhält man so ein Alkylhalogenid bzw. Halogenalkan.
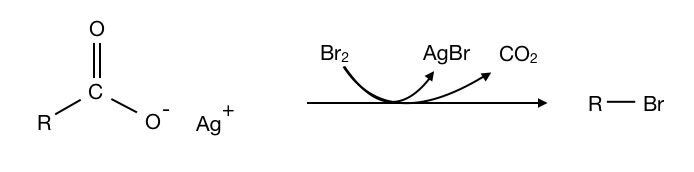
Die Hunsdiecker-Reaktion
Quellen:
- Morrison, Boyd, Organic Chemistry, 7th Edition 2011
- Römpp Chemie-Lexikon, 9. Auflage 1992
- Wikipedia, Artikel zu den einzelnen Säuren
- Chemgapedia, Artikel "Reaktion von Carbonsäuren"
- Wikipedia, Artikel "Malonsäure"