Die Struktur eines größeren Proteins lässt sich auf vier Ebenen mit zunehmender Komplexität beschreiben:
- Primärstruktur
- Sekundärstrukturen
- Tertiärstruktur
- Quartärstruktur
Die einfachste, erste oder unterste Ebene ist die Primärstruktur, die wir auf dieser Seite näher betrachten wollen.
Ein Beispiel
Gehen wir einmal zurück in das Jahr 1953. In diesem Jahr wurde die Aminosäuresequenz des Insulins aufgeklärt, und zwar von Frederick Sanger (1918-2013). Sanger erhielt dafür 1958 den Nobelpreis für Chemie. Sanger ist übrigens einer der wenigen Forscher, die zwei Nobelpreise erhielten. 1980 bekam er den zweiten Nobelpreis für Chemie, diesmal für die Aufklärung der Basensequenz in Nucleinsäuren.
Das Hormon Insulin, das für die Regulation des Blutzuckerspiegels verantwortlich ist, besteht aus zwei Peptidketten, die über Disulfidbrücken fest miteinander verbunden sind. Schauen wir uns die Aminosäuresequenz der langen Insulinkette an:

Die Primärstruktur der langen Insulin-Kette
Autor: Ulrich Helmich, Lizenz: Public domain.
Die lange Kette des Insulins, die sogenannte B-Kette, besteht aus 30 Aminosäuren. Das Amino-Ende der B-Kette wird von der Aminosäure Phenylalanin gebildet, und das Carboxy-Ende besteht aus der Aminosäure Threonin. Die A-Kette, im Bild nicht zu sehen, enthält nur 21 Aminosäuren.
Die Reihenfolge der Aminosäuren in einer Peptidkette, die Aminosäuresequenz, ist genetisch festgelegt und wird meistens als Primärstruktur bezeichnet.
Primärstruktur
Die genetisch festgelegte Reihenfolge der Aminosäuren in einem Peptid oder Protein. Man spricht auch oft von einer Aminosäuresequenz.
Die Primärstruktur ist genetisch festgelegt
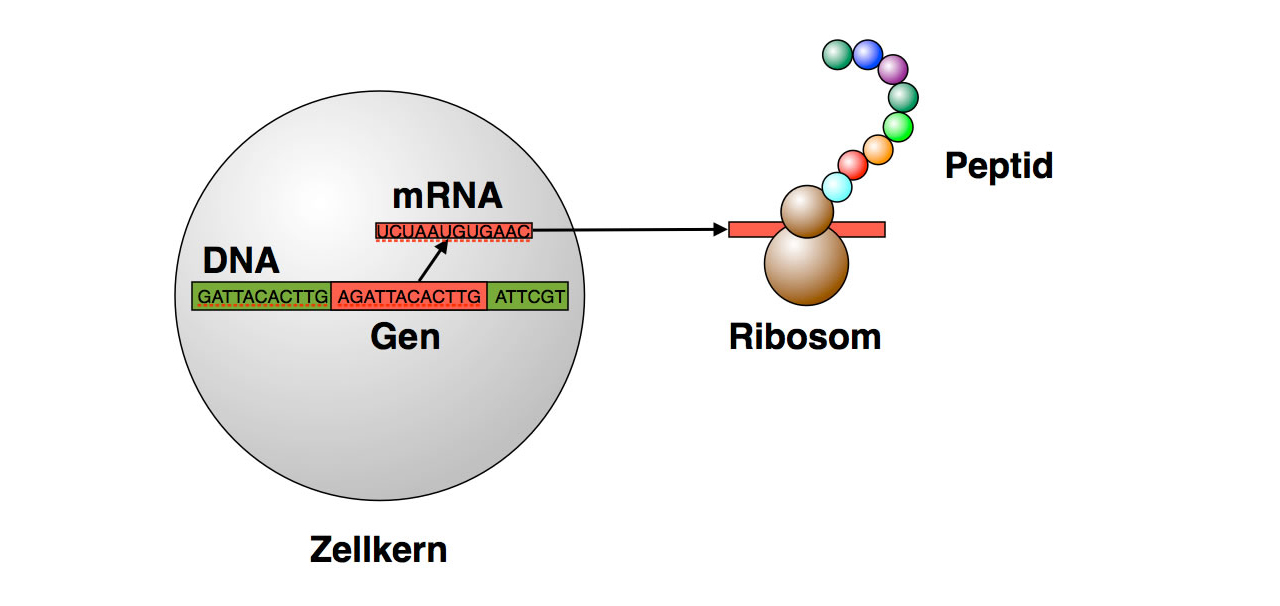
Die Aminosäure-Sequenz ist in der DNA festgelegt
Autor: Ulrich Helmich, Lizenz: Public domain.
Peptide und Proteine werden an den Ribosomen der Zelle hergestellt, der Vorgang wird als Translation bezeichnet (= "Übersetzung"). Die Ribosomen müssen die Aminosäuren in der richtigen Reihenfolge aneinander heften. Die dafür notwendige Information beziehen die Ribosomen aus dem Zellkern in Form einer messenger-RNA (mRNA, Boten-RNA). Die mRNA wiederum ist eine Kopie eines DNA-Abschnitts. Die Herstellung dieser Kopie im Zellkern wird als Transkription (= "Abschrift") bezeichnet. DNA-Abschnitte, die Informationen zum Bau eines Proteins enthalten, bezeichnet man als Gene.
Sekundär- Tertiär- und Quartärstrukturen
Bereits in der Reihenfolge der Aminosäuren ist die spätere tatsächliche Raumstruktur eines Proteins festgelegt. Schon während der Translation faltet sich die Peptidkette zu spiraligen oder blattähnlichen Strukturen, den sogenannten α-Helices und β-Faltblättern. Solche Sekundärstrukturen sind energetisch günstiger als die langgestreckte Peptidkette, weil durch die Bildung von Sekundärstrukturen hydrophobe Seitenketten besser vor dem wässrigen Medium geschützt werden können.
Die Sekundärstrukturen wiederum können sich zu komplexen Tertiärstrukturen auffalten, die durch schwache chemische Bindungen, durch Ionenbindungen und durch Disulfidbrücken zusammengehalten werden.
Bei der Bildung der Sekundärstrukturen und Tertiärstrukturen sind oft bestimmte Enzyme beteiligt, die sogenannten Chaperone. Manche dieser Strukturen können sich aber auch von selbst bilden, weil sie energetisch günstiger sind.
Viele größere Proteine bestehen aus mehreren Untereinheiten, die selbst wieder eine Tertiärstruktur haben. Bei solchen aus mehreren Untereinheiten zusammengesetzten Proteinen spricht man dann von einer Quartärstruktur.
Aufklärung von Primärstrukturen
Kommen wir wieder auf Frederick Sanger (1918-2013) zurück, der die Primärstruktur des Insulins aufgeklärt und damit wichtige Pionierarbeit geleistet hat. Wie kann man die Primärstruktur eines Proteins aufklären, wenn man die Basensequenz des zuständigen Gens nicht kennt?
Wenn man die Basensequenz des verantwortlichen Gens kennt, ist es kein Problem, aus der Reihenfolge der mRNA-Basentripletts die Reihenfolge der Aminosäuren im Protein zu berechnen. Jedes Basentriplett auf der mRNA codiert eine Aminosäure. Der Code GAG auf der mRNA steht beispielsweise für die Aminosäure Glutaminsäure.
Die Struktur der DNA wurde aber ebenfalls erst im Jahre 1953 von Watson und Crick aufgeklärt, und bis zur Entschlüsselung des genetischen Codes vergingen noch ein paar Jahre. Sanger konnte also nicht auf die Basensequenz des Insulingens zurückgreifen, sondern musste die Aminosäuresequenz des Hormons in mühseliger Kleinarbeit mit chemischen Methoden aufklären.
Edman-Abbau
Bei diesem Verfahren, das heute nicht mehr eingesetzt wird, markiert man zunächst die Aminosäure am N-Terminus des Proteins. Das geht recht einfach, weil dies die einzige Aminosäure des Proteins ist, die eine unveränderte NH2-Gruppe besitzt. Zur Markierung wird die Verbindung 1-Fluor-2,4-dinitrobenzol eingesetzt.
Anschließend wird das Protein durch Proteasen in kurze Peptide und Aminosäuren zerlegt, und man trennt das Peptid/Aminosäure-Gemisch mit Verfahren wie Elektrophorese in seine Bestandteile auf. Nun kann man die markierte Aminosäure leicht identifizieren.
Eine weitere Probe des Proteins wird dann mit einer Amino-Exopeptidase behandelt, welche die Aminosäure am N-Terminus des Proteins abspaltet. Dann markiert man die neue N-terminale Aminosäure und verfährt wie oben beschrieben. Das Ganze wiederholt man solange, bis man alle Aminosäuren identifiziert hat.
Modernere Methoden
Insgesamt ist das Edman-Verfahren sehr zeitaufwendig und teuer und wird daher heute nicht mehr angewandt. "Um die Aminosäuresequenz eines Proteins zu bestimmen, wird meist entweder die Gensequenz der DNA aus einer DNA-Sequenzierung oder aus einer Datenbank sequenzierter Genome wie tBLAST in silico in eine Proteinsequenz übersetzt". [3]
Auch die Methode der Massenspektrometrie wird zunehmend für die Identifizierung von Proteinen eingesetzt. Jedes Protein hat eine charakteristische Masse, und ein Vergleich mit in großen Datenbanken gespeicherten Werten liefert oft schon eine eindeutige Identifizierung des Proteins oder Peptids.
Quellen:
- Römpp Chemie-Lexikon, 9. Auflage 1992
- Alberts, Bruce et al. Molekularbiologie der Zelle, 6. Auflage, Weinheim 2017.
- Wikipedia, Artikel Edman-Abbau.
Seitenanfang -
Weiter mit den Sekundärstrukturen...